ARTIGO
ORIGINAL
Associação
entre pico de fluxo da tosse, colonização bacteriana e estado nutricional em
crianças e adolescentes com fibrose cística
Association of cough peak flow with chronic bacterial colonization and
nutritional status in children and adolescents with cystic fibrosis
Bruna Milaine Broedel Vaz*, Jéssica
Barbosa Falcão*, Fernanda Mayrink Gonçalves Liberato, Ft.,D.Sc.**,
Luana da Silva Baptista Arpini, M.Sc.***,
Veronica Lourenço Wittmer, D.Sc.****,
Flávia Marini Paro, Ft., D.Sc.*****
*Graduada
em Fisioterapia pela Universidade Federal do Espírito Santo, Departamento de
Educação Integrada em Saúde, Centro de Ciências da Saúde, Universidade Federal
do Espírito Santo, Vitória/ES, **Fisioterapeuta do Hospital Estadual Infantil
Nossa Senhora da Glória, Vitória/ES, ***Nutricionista do Hospital Estadual
Infantil Nossa Senhora da Glória, Vitória/ES, ****Docente do Departamento de
Educação Integrada em Saúde, Centro de Ciências da Saúde, Universidade Federal
do Espírito Santo, *****Docente do Departamento de Educação Integrada em Saúde,
Centro de Ciências da Saúde, Universidade Federal do Espírito Santo
Recebido em 12 de
abril de 2018; aceito em 26 de setembro de 2018.
Endereço
de correspondência:
Flávia Marini Paro, Universidade Federal do Espírito Santo (UFES), Centro de
Ciências da Saúde, Departamento de Educação Integrada em Saúde, Av. Mal.
Campos, 1468 Maruípe 29043-900 Vitória ES, E-mail:
flamarp@yahoo.com; Bruna Milaine Broedel
Vaz: brunavaz14@gmail.com; Jéssica Barbosa Falcão: jessicabarbosafalcao@hotmail.com;
Fernanda Mayrink Gonçalves Liberato: nandamayrink@yahoo.com.br; Luana da Silva
Baptista Arpini: luanaarpini@hotmail.com; Veronica
Lourenço Wittmer: ve_lourenco@yahoo.com.br
Resumo
Introdução: Este estudo teve
como objetivo avaliar a associação entre pico de fluxo da tosse (PFT),
colonização bacteriana crônica e estado nutricional em
crianças e adolescentes com Fibrose Cística. Métodos: Estudo transversal, com amostra por conveniência composta
por indivíduos com FC (7-18 anos), cadastrados em um hospital de referência
estadual. Foi avaliado o PFT por um medidor portátil de pico de fluxo
expiratório. Dados clínicos, antropométricos e sobre colonização bacteriana
foram colhidos nos prontuários. O estado nutricional foi classificado pelo
percentil do índice de massa corporal para a idade. Para a análise estatística
foram usados os testes: Shapiro-Wilk, Exato de
Fisher, Teste T Student independente, Mann-Whitney e
Correlação de Spearman. Foi considerado significante
p<0,05. Resultados: Na
caracterização da amostra, houve predomínio das seguintes características:
redução do PFT (82,35%), risco nutricional (70,6%), colonização bacteriana
crônica (82,4%). Indivíduos colonizados por Pseudomonas aeruginosa apresentaram maior percentual
de redução do PFT (p=0,045). Conclusão:
O pico de fluxo da tosse apresentou-se reduzido nas crianças e adolescentes com
FC da amostra estudada, sendo essa redução mais acentuada nos indivíduos
colonizados por Pseudomonas aeruginosa do
que naqueles colonizados por Staphylococcus aureus.
A maioria dos indivíduos apresentava-se em risco nutricional, mas não foi
observada correlação entre estado nutricional e pico de fluxo da tosse.
Palavras-chave: avaliação
nutricional, infecção, fibrose cística, tosse, pseudomonas
aeruginosa.
Abstract
Introduction: The aim of this study was to evaluate the association among cough peak
flow (CPF), chronic bacterial colonization and nutritional status in children
and adolescents diagnosed with cystic fibrosis. Methods: A cross-sectional study with a convenience sample of
individuals diagnosed with CF (7-18 years old) enrolled in a referral hospital
of a Brazilian State. The CPF was evaluated by a portable expiratory peak flow
meter. Clinical, anthropometric and bacterial colonization data were collected
in the medical records. Nutritional status was classified according to the
percentile of the body mass index for age. Statistical analysis was performed
using the following tests: Shapiro Wilk, Fisher's
Exact, Student's t, Mann-Whitney and Spearman's Correlations, being considered
significant p<0.05. Results: The
following characteristics predominated: CPF reduction (82.35%), nutritional
risk (70.6%), chronic bacterial colonization (82.4%).
Individuals colonized by Pseudomonas aeruginosa had a higher percentage of CPF reduction
(p=0.045). Conclusion: The cough peak
flow was reduced in the children and adolescents with cystic fibrosis in the
studied sample, and this reduction was more pronounced in individuals colonized
by Pseudomonas aeruginosa
than in those colonized by Staphylococcus
aureus. Most individuals were at nutritional
risk, but no correlation was observed between nutritional status and cough peak
flow.
Key-words: nutrition
assessment, infection, cystic fibrosis, cough, pseudomonas aeruginosa.
Introdução
A
Fibrose Cística
(FC) é uma doença genética autossômica
recessiva, caracterizada por um
desequilíbrio do transporte iônico, que provoca o aumento
da viscosidade das
secreções, causando obstrução dos ductos
das glândulas, inflamação, lesão e
destruição tecidual progressiva, com consequências multissistêmicas
[1].
No sistema
respiratório, a secreção espessa e viscosa causa obstrução das vias aéreas
distais, que leva à doença pulmonar obstrutiva crônica, podendo causar bronquiectasias desde os primeiros meses de vida, infecções
recorrentes e colonização por bactérias e outros microrganismos. A principal
causa de morbidade e mortalidade da doença é a deterioração progressiva da
função pulmonar [2].
Além disso, os
indivíduos são, em sua maioria, acometidos por insuficiência pancreática, uma
importante causa de desnutrição nesses pacientes. O mau funcionamento do
pâncreas exócrino causa comprometimento nutricional devido à má absorção e esteatorréia, comuns desde o nascimento. A desnutrição
contribui para o déficit de crescimento [3] e está relacionada ao declínio da
função pulmonar [3,4], e da qualidade de vida [5].
Evidências mostraram
que a colonização por Pseudomonas aeruginosa
está associada à redução função pulmonar [6] e tem impacto negativo no
prognóstico desses pacientes [6,7], sendo considerada o
principal preditor de mortalidade em crianças
com FC [8]. Sendo assim, é fundamental prevenir e tratar as infecções
precocemente, visando reduzir complicações e adiar colonizações.
A tosse é um
mecanismo fisiológico necessário para a prevenção de infecções, por seu papel
fundamental na remoção das secreções e higiene brônquica, sendo importante
avaliar sua eficácia [9]. O uso de um dispositivo portátil, de baixo custo e
não invasivo para medir o pico de fluxo da tosse (PFT) fornece dados funcionais
importantes, e um meio para analisar os resultados das intervenções que visem
melhorar a tosse. Além disso, seu uso pode economizar tempo e reduzir custos
[10].
O PFT avalia a
efetividade da tosse de forma simples e reprodutível [9-11], tendo utilização
crescente na prática clínica e em estudos científicos [9-13]. Entretanto, foi
pouco estudado na FC [14], sendo necessárias mais pesquisas, que poderão
favorecer a prevenção e o tratamento precoce de infecções e comorbidades
influenciadas pela ineficácia da tosse.
O objetivo deste
estudo foi avaliar a associação do PFT com a colonização crônica por bactérias
e com o estado nutricional em crianças e adolescentes com FC.
Material
e métodos
Estudo descritivo
transversal, cuja coleta de dados foi realizada entre julho a dezembro de 2016,
no centro de referência estadual infantil para o tratamento da FC do Espírito
Santo. O estudo foi aprovado pelo Comitê de Ética em Pesquisa da Universidade
Federal do Espírito Santo (Parecer no 352/572, CAAE: 16947313.1.0000.5060) e
pelo Comitê de Ética em Pesquisa do Hospital Infantil Nossa Senhora da Glória
(Parecer: 368/920, CAAE: 16947313.1.0000.5060), sendo conduzido em todas as
suas etapas de acordo com a Resolução do CNE 466/12, que regulamenta as pesquisas
envolvendo seres humanos no Brasil.
Os critérios de
inclusão foram: pacientes com diagnóstico de FC, na faixa etária de 7 a 18
anos, cadastrados no centro de referência e atendidos pelo serviço de
fisioterapia no período da coleta de dados. O critério diagnóstico de FC
utilizado foi o teste do suor positivo ou teste de suor duvidoso com
identificação de mutações no gene cystic fibrosis transmembrane regulator (CFTR) condizentes com o quadro clínico de FC
[15,16]. Os critérios de exclusão foram: pacientes com doenças associadas que
prejudicassem a compreensão e/ou a execução dos testes, incluindo exacerbação
do quadro pulmonar; pacientes não atendidos pela fisioterapia nos dias das
coletas de dados e pacientes que se recusassem a participar ou a assinar o
termo de consentimento livre e esclarecido (TCLE), após todas as informações e
esclarecimentos de dúvidas sobre o projeto. No caso dos menores de 18 anos,
além do paciente, o responsável também precisavam concordar
e assinar o TCLE, conforme resolução CNE 466/12.
Havia um total 48
pacientes cadastrados, com diagnóstico de FC, na faixa etária do estudo, dos
quais 26 não estiveram presentes em nenhum dos dias nos quais foram coletados
os dados, não sendo, portanto, incluídos no estudo. Foram excluídos dois
indivíduos por autismo, um indivíduo por neuropatia, um indivíduo que não
consentiu em participar e um indivíduo que não pode realizar o teste devido à
exacerbação do quadro pulmonar. A amostra por conveniência foi constituída de
17 participantes.
Após a assinatura do
TCLE, foi preenchida uma ficha de avaliação contendo informações demográficas e
clínicas. As informações antropométricas e sobre colonização crônica por
bactérias foram coletadas dos prontuários.
Foi avaliado o PFT
utilizando o dispositivo portátil de medição do pico de fluxo expiratório (PFE)
Medicate Peak Flow Meter (Fyne Dynamics Ltd, Harlow, Inglaterra), com
variação de 60 – 900 l/min. Para a realização do teste de PFT, o paciente
permanecia sentado numa cadeira sem braços, e era orientado a manter a boca
completamente acoplada no bucal, para evitar escape de ar durante a execução do
teste. Era pedida uma inspiração máxima, seguida de uma tosse voluntária
(expiração rápida, curta e explosiva), realizando um mínimo de três repetições
[12]. Caso houvesse diferença entre as três primeiras tentativas
maior que 10% [17,18] ou 20 l/min [17], permitia-se a realização do
teste em até 10 tentativas, utilizando o maior valor como resultado [17,18].
Para a avaliação dos
resultados, foram usados os valores de referência publicados no estudo de
Bianchi e Baiardi, realizado na Itália com crianças e
adolescentes saudáveis [19], tendo em vista que não existem estudos com a
população brasileira nesta faixa etária.
O critério utilizado
para avaliação da variação do PFT em relação à normalidade foi baseado na
Diretriz para Testes de Função Pulmonar da Sociedade Brasileira de Pneumologia
e Tisiologia (SBPT) [17], que considera o quinto percentil como o melhor
critério para limite inferior de normalidade dos parâmetros espirométricos,
incluindo o PFE.
As variações do PFT
em relação à normalidade foram calculadas baseadas na coluna de quinto
percentil do estudo de Bianchi e Baiardi do qual foi
subtraído o maior valor do PFT do indivíduo, e a partir desse resultado foi
calculado o percentual de redução em relação ao quinto percentil dos valores de
referência [19].
Para a avaliação do
estado nutricional, dados de antropometria (massa corporal e altura) foram
obtidos para cálculo do índice de massa corporal (IMC) e determinação do
percentil do índice de massa corporal para idade (IMC/I), que foi calculado
utilizando o software WHO Anthro Plus, 2007 versão 1.0.4. A classificação do
estado nutricional seguiu o ponto de corte recomendado por Turck
et al. [20]: "eutrofia”-
IMC/I ≥ percentil 50 e “risco nutricional”- IMC/I < percentil
50.
Para análise
estatística foi utilizado o software Statistical Package for Social Sciences
(SPSS) versão 22 (IBM, Armonk, NY, Estados Unidos).
Nas análises descritivas as variáveis categóricas foram apresentadas em
frequências absolutas e relativas. O teste de Shapiro-Wilk
foi aplicado para avaliar se os dados apresentavam distribuição normal. As
variáveis paramétricas e não paramétricas foram apresentadas, respectivamente,
como média e desvio padrão (DP) ou como mediana e intervalo interquartílico
(IIQ). Para o cruzamento entre duas variáveis categóricas foi usado o teste
Exato de Fisher. Os dados paramétricos foram analizados
usando-se o Teste t Student independente. O Teste de
Mann-Whitney foi usado para os testes não paramétricos. Para a análise de
correlação foi utilizada a Correlação de Spearman
(não paramétrica). Para avaliar a força das correlações, foi considerado o
seguinte critério [21]: se 0 < r < 0,3: fraca; se 0,3 ≤ r < 0,6:
moderada; se 0,6 ≤ r < 0,9: forte; se 0,9 ≤ r < 1: muito
forte. Foi considerado significante p < 0,05.
Resultados
A população do estudo
foi constituída por 17 indivíduos com diagnóstico de FC, com média de 12,35 ±
3,62 anos de idade, variando de 7 a 18 anos. A caracterização antropométrica e
demográfica da amostra, bem como os valores médios de PFT e sua variação em
relação aos valores de referência estão detalhados na tabela I, na qual é
possível observar que não houve diferença entre os gêneros com relação à
distribuição dessas características.
A maioria dos
pacientes (n = 14; 82,4%) apresentou variação negativa do PFT em relação ao
quinto percentil dos valores de referência, ou seja, os resultados do PFT
estavam reduzidos em relação ao quinto percentil, variando esta redução de
-1,16% a -45,34%, o que indica que o PFT nesses indivíduos pode ser considerado
abaixo da normalidade (Tabela I). Apenas um indivíduo apresentou valor igual ao
quinto percentil e dois indivíduos apresentaram variação percentual positiva em
relação ao quinto percentil, sendo que nenhum deles atingiu o décimo percentil
dos valores de referência.
Tabela
I - Variáveis antropométricas, demográficas e
pico de fluxo da tosse em indivíduos com fibrose cística e resultados dos
testes de comparação entre os gêneros.
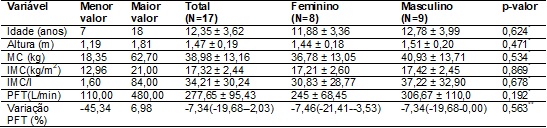
Dados apresentados em
média ± desvio padrão ou mediana (intervalo interquartílico); N = número de
indivíduos; MC = massa corporal; IMC = índice de massa corporal; IMC/I =
percentil do índice de massa corporal para a idade; Variação PFT = redução ou
aumento do pico de fluxo da tosse em relação ao quinto percentil dos valores de
referência; PFT = pico de fluxo da tosse; *Teste t Student Independente; **Teste de Mann-Whitney; Considerado
significante p < 0,05.
Houve maior
prevalência de indivíduos da raça branca (n=11; 64,7%) e com risco nutricional
(n=12; 70,6%), considerando-se ponto de corte recomendado por Turck et al., que classifica todos
os indivíduos com IMC/I< percentil 50 como risco nutricional [20]. A maioria
dos indivíduos (n=14; 82,4%) era colonizada por bactérias (Tabela II).
Tabela
II –
Distribuição entre os gêneros das variáveis raça, estado nutricional e colonização bacteriana
nos indivíduos com fibrose cística (7 a 18 anos.
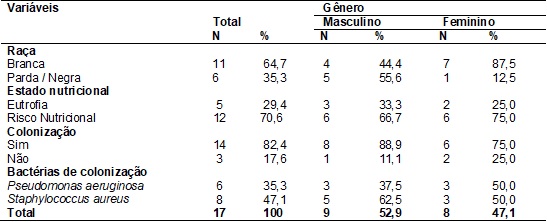
N = número de
indivíduos
Foi encontrada
correlação positiva entre o PFT e as seguintes variáveis: idade, massa
corporal, altura e IMC. Não foi encontrada correlação entre o PFT e o IMC/I
percentil. Também não foi observada correlação entre a variação do PFT e essas
variáveis (tabela III).
Tabela
III
– Correlações entre o pico de fluxo da
tosse e as variáveis antropométricas em indivíduos com fibrose cística.
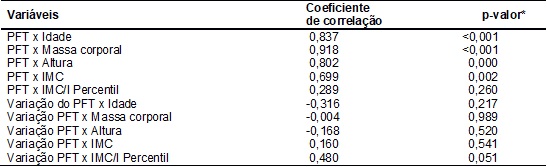
PFT = pico de fluxo da tosse; IMC = índice de massa corporal;
IMC/I Percentil = percentil do índice de massa corporal para a idade; Variação
PFT = redução ou aumento do PFT em relação ao quinto percentil dos valores de
referência; Coeficiente de Correlação de Spearman;
*considerado significante p < 0,05.
Não houve diferença
significativa na comparação dos pacientes colonizados por Pseudomonas
aeruginosa e Staphylococcus
aureus em relação às variáveis: idade, altura, massa corporal, IMC, e PFT
(Tabela IV).
Quando comparou-se a variação do PFT entre os pacientes colonizados
por Pseudomonas aeruginosa e
os pacientes colonizados por Staphylococcus aureus,
verificamos diferença significativa (p=0,045), podendo-se dizer que os
indivíduos com Pseudomonas aeruginosa
tiveram maior variação negativa do PFT em relação aos valores de referência do
que os indivíduos com Staphylococcus aureus. Devido ao número reduzido de
pacientes não colonizados, apenas três indivíduos, não foi possível aplicar
testes estatísticos para a comparação entre os 3
grupos, sendo assim, o grupo de indivíduos não colonizados não foi incluído nos
testes de comparação. Sendo assim, as comparações foram feitas apenas entre o
grupo colonizado com Pseudomonas aeruginosa e
o grupo colonizado por Staphylococcus aureus (Tabela IV).
Tabela
IV –
Distribuição das variáveis antropométricas
e dos valores de pico de fluxo da tosse segundo o estado de colonização
bacteriana dos indivíduos com fibrose cística (07 a 18 anos) e comparações
entre os grupos.
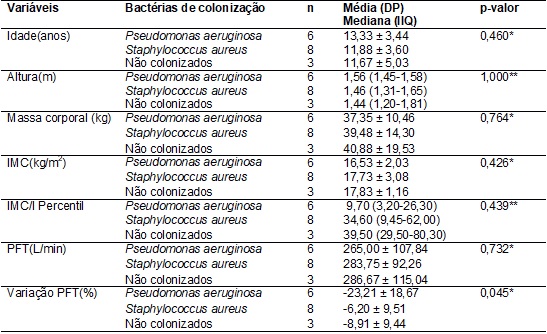
Dados apresentados em
média ± desvio padrão ou mediana (intervalo interquartílico). IMC = índice de
massa corporal; IMC/I Percentil = percentil do índice de massa corporal para a
idade; PFT = pico de fluxo de tosse; Variação PFT =
redução ou aumento do PFT em relação ao quinto percentil dos valores de
referência; *Teste t Student independente; **Teste de
Mann-Whitney; considerado significante p < 0,05.
Discussão
Os resultados
mostraram que a maioria das crianças e adolescentes com FC apresentou PFT
reduzido em relação ao quinto percentil dos valores de referência [19], o que
pode ser considerado abaixo da normalidade [17]. Além disso, esta redução foi
maior no grupo de crianças e adolescentes colonizados por Pseudomonas aeruginosa em comparação com o grupo
colonizado por Staphylococcus aureus, o que ainda não havia sido
observado em estudos anteriores.
Devido à inexistência
de estudos com valores de referência do PFT para crianças brasileiras na faixa
etária estudada, foram usados valores de referência de um estudo realizado na
Itália com crianças e adolescentes saudáveis [19]. O único estudo encontrado
sobre o PFT na FC avaliou indivíduos com média de idade de 26 ± 10 anos, o que
impossibilita a comparação com nossos resultados, embora também tenha sido
observado PFT reduzido para a faixa etária estudada [12].
Diversas pesquisas
têm mostrado que a colonização por Pseudomonas aeruginosa
apresenta relação com a deterioração mais rápida da função pulmonar nos
indivíduos com FC [8,22-25], inclusive, quando comparada à colonização por Staphylococcus aureus [25]. Um estudo que avaliou, ao
longo de 25 anos, o declínio da função pulmonar de 16.619 indivíduos com FC
(incluindo crianças), atendidos por 117 centros de referência dos Estados
Unidos, concluiu que os indivíduos colonizados por Pseudomonas aeruginosa apresentam maior declínio
anual da função pulmonar, avaliada no estudo pelo volume expiratório forçado no
primeiro segundo (VEF1), do que os indivíduos colonizados por Staphylococcus aureus [25].
Embora a função
pulmonar não tenha sido avaliada no presente estudo, é possível supor que a
maior redução do PFT observada nos indivíduos com Pseudomonas aeruginosa, esteja relacionada ao maior
comprometimento da função pulmonar, que seria esperado nesses pacientes
[8,22-25]. Essa suposição pode ser feita com base nas evidências que demonstram
correlação positiva entre o PFT e o VEF1 [19,26] e entre a redução do PFT e o
declínio anual do VEF1 na FC [14]. O estudo de Bianchi e Baiardi
[19], com crianças saudáveis na faixa etária do presente estudo, mostrou que o
PFT apresentou forte correlação com as seguintes variáveis espirométricas:
capacidade vital forçada (CVF), VEF1 e PFE. Do ponto de vista fisiológico, esta
correlação é esperada, considerando-se que o PFT depende, entre outros fatores,
da capacidade de gerar fluxos expiratórios adequados [9,27], sendo tal
capacidade comprometida nas doenças obstrutivas crônicas [9], como a FC, nas
quais a redução do fluxo expiratório e o aumento na viscosidade da secreção são
considerados as principais causas de inefetividade da
tosse [9].
Para elucidar o papel
das infecções respiratórias na FC, têm sido estudadas as interações entre os
mediadores inflamatórios e os diferentes microrganismos, que, nesta doença,
propiciam um microambiente inflamatório tóxico no sistema respiratório [2,28].
As secreções espessas e viscosas comprometem a eficiência do transporte mucociliar na proteção das vias aéreas. Além disso, o
desequilíbrio hidroeletrolítico causa alterações no pH
local, o que prejudica a função dos peptídeos antimicrobianos e das células de
defesa contra os microrganismos patogênicos [2]. Tais alterações têm como
consequência uma resposta inflamatória exagerada, ineficaz e persistente, que
provoca danos estruturais permanentes nas vias aéreas e no parênquima pulmonar
[2,24], bronquiectasias desde a primeira infância
[2], doença pulmonar obstrutiva crônica e insuficiência respiratória [2,24,28].
Crianças com FC são,
em geral, infectadas precocemente por bactérias gram-positivas, o que resulta
em resposta inflamatória exacerbada e danos às vias aéreas, aumentando a
suscetibilidade à colonização por bactérias gram-negativas como a Pseudomonas aeruginosa
[2], considerada um dos mais importantes preditores
de morbidade e mortalidade na FC [8,29]. A colonização crônica por Pseudomonas aeruginosa
deve-se principalmente às estirpes mucoides produtoras de um biofilme
bacteriano que aumenta a tolerância aos antibióticos, resiste à fagocitose e é
relativamente impermeável aos componentes do sistema imunológico inato e
adaptável [29].
Por todas essas
razões, a eficiência da tosse e a otimização da
higiene brônquica são muito importantes nesses indivíduos, e as mais recentes
diretrizes nacionais [16] e internacionais [29-31] para o tratamento de FC
preconizam que seja enfatizada a prevenção das infecções e colonizações, o que
deve incluir a fisioterapia desde o diagnóstico e por toda a vida [16,29-31],
associada ao uso de nebulizações, agentes mucolíticos
e antimicrobianos [16,29-31], pois os benefícios da fisioterapia para estes
pacientes estão bem evidenciados na literatura [16,29-33].
A média de idade dos
indivíduos colonizados por Pseudomonas aeruginosa encontrada neste estudo (13,33 ±3,44 anos)
corrobora dados recentes da literatura, que considera a faixa etária mais
frequente de início da colonização por esta bactéria entre 10 e 20 anos de
idade [2]. Outros estudos mostraram que indivíduos do sexo feminino apresentam
maior prevalência de infecção por Pseudomonas aeruginosa
[8,34], e risco aumentado de colonização por
esta bactéria em idades mais precoces do que os
indivíduos do sexo masculino
[34]. No presente estudo, não foi possível fazer a
comparação estatística entre
os gêneros com relação à
colonização por bactérias, devido ao número
amostral
reduzido para essas categorizações, contudo, vale a pena
salientar que 50% das
meninas e 37,5% dos meninos apresentavam colonização por Pseudomonas aeruginosa, o que mostra uma tendência
de maior prevalência de colonização nas meninas, que talvez pudesse ser
confirmada com um número amostral maior.
Os resultados
mostraram alta prevalência de risco nutricional na amostra (70,6%), o que é
frequente nas crianças e adolescentes com FC [3-5,20,32,35].
Não houve diferença significativa entre os pacientes colonizados por Pseudomonas aeruginosa e Staphylococcus aureus, em relação às variáveis
nutricionais, o que corrobora um estudo longitudinal recente, realizado no
Brasil, que mostrou que o estado nutricional não foi fator de risco para
infecção por P. aeruginosa
[36]. A variação do PFT não apresentou correlação com o percentil do IMC
para a idade, sugerindo que na amostra estudada não houve relação entre o
estado nutricional e o PFT. Está bem estabelecida na literatura, a relação entre desnutrição e função pulmonar na FC [3,4],
sendo assim, é importante a realização de outros estudos, com maior número
amostral, que avaliem a relação do PFT com o estado nutricional de crianças e
adolescentes com esta doença.
Foram observadas
correlações entre o PFT e as seguintes variáveis antropométricas: idade,
altura, massa corporal e IMC, corroborando os resultados de Bianchi e Baiardi [19], que mostraram correlação do PFT com as mesmas
variáveis nesta faixa etária. As correlações do PFT com a idade e a altura já
eram esperadas, pois estão bem descritas na literatura, sendo a idade inclusive
usada nas tabelas de referência para PFT em crianças e adolescentes [19,27].
Outra publicação mostrou que o peso e a altura estavam pouco associados à
função pulmonar aos três anos de idade, mas fortemente associados aos seis anos
[37], o que também está de acordo com os resultados deste estudo, cuja amostra
foi constituída por indivíduos com idade a partir de 7
anos.
A redução do PFT
observada no presente estudo reforça a importância de que esta avaliação seja
realizada nos indivíduos com FC, pois a tosse é um mecanismo fundamental para a
prevenção de infecções [9,38] e deve ser monitorada nas avaliações e
reavaliações desses pacientes. Tem sido evidenciada boa reprodutibilidade da
avaliação do PFT por meio do medidor analógico portátil de pico de fluxo
expiratório [10,11], inclusive para crianças a partir de 4
anos [11], e boa acurácia desse tipo de medidor em comparação com o pneumotacógrafo, considerado padrão ouro para essa medida
[10]. Apenas uma publicação relatou inacurácia do
aparelho portátil em comparação com o pneumotacógrafo
[39]. Com um método fácil e de baixo custo para o monitoramento do PFT é
possível planejar intervenções que contribuam para a melhora desta variável
[27], sendo também importante a realização de estudos que busquem evidências
sobre as intervenções que tenham impacto positivo no PFT e em outras variáveis espirométricas em pacientes com FC, como um estudo recente
que mostrou que o método Pilates melhora o PFE em
pacientes com doença pulmonar obstrutiva crônica [33].
As principais
limitações do estudo foram o número reduzido de participantes e a amostra de
conveniência, que impossibilitam a generalização dos resultados. Contudo,
segundo o Registro Brasileiro de FC [40], o total de pacientes cadastrados e em
tratamento para FC no ES, incluindo adultos e crianças era 135 no último
relatório, e o estudo foi realizado no único centro de referência pediátrico do
ES para o tratamento da FC, para o qual são encaminhados pacientes de todos os
municípios do ES após a triagem neonatal ou o diagnóstico de FC [35,40]. O
centro de referência tinha, no período da coleta de dados, 48 pacientes
cadastrados na faixa etária estudada, sendo assim, embora pequena, a amostra
corresponde a aproximadamente 35,42% dos indivíduos com FC do ES, nesta faixa
etária.
Outra limitação do
estudo, foi a falta de valores de referência
brasileiros para o PFT em crianças e adolescente na faixa etária estudada.
Contudo, devemos salientar que um estudo recente avaliou o PFT em crianças
brasileiras saudáveis em idade pré-escolar (4 a 6 anos) [11] e encontrou, para
esta faixa etária, resultados dentro da faixa de normalidade dos valores
descritos por Bianchi e Baiardi [19], o que reforça a
possibilidade de utilização dos mesmos como parâmetros no presente estudo.
Conclusão
O PFT apresentou-se
reduzido nas crianças e adolescentes com FC da amostra estudada, sendo essa
redução mais acentuada nos indivíduos colonizados por Pseudomonas aeruginosa do que nos indivíduos
colonizados por Staphylococcus aureus.
Com relação ao estado
nutricional, houve alta prevalência de indivíduos que apresentavam risco
nutricional, mas não foi encontrada correlação entre o PFT e o estado nutrional, e também não foi observada associação entre o
estado nutricional e a colonização bacteriana.
Considerando que a
colonização por Pseudomonas aeruginosa é
um dos mais importantes preditores de mortalidade na
FC e que a tosse é um mecanismo fisiológico fundamental para a prevenção de
infecções respiratórias, são necessários mais estudos sobre a associação dessas
variáveis em pacientes com FC, pois a literatura é escassa com relação a este
assunto, bem como sobre valores de referência para o PFT em crianças e
adolescentes brasileiros.
A redução do PFT
observada no presente estudo também evidencia a importância de que esta
variável seja avaliada e monitorada ao longo do tempo nos pacientes com fibrose
cística, o que pode contribuir para o planejamento de intervenções direcionadas
para prevenir a redução da eficácia da tosse nesses pacientes.
Referências
- Cutting GR. Cystic fibrosis genetics: from molecular understanding to
clinical application. Nature Reviews Genetics 2015;16(1):45-56.
- Elborn JS. Cystic
fibrosis. Lancet 2016;388:2519-31.
- Hortencio TDR, Nogueira RJN, Marson FAL, Hessel G, Ribeiro JD, Ribeiro AF. Fatores que afetam o crescimento e estado
nutricional de pacientes com fibrose cística com idade inferior a 10 anos e que
não foram submetidos à triagem neonatal. Rev Paul Pediatr 2015;33(1):3-11.
- Mauch RM, Kmit AHP, Marson FAL, Levy CE,
Barros-Filho AA, Ribeiro JD. Associação dos parâmetros de crescimento e
nutricionais com função pulmonar na fibrose cística: revisão da literatura. Rev Paul Pediatr 2016;34(4):503-9.
- Silva LA, Lima ACP, Wittmer VL, Liberato FMG, Arpini
LSB, Paro FM. Qualidade de vida de crianças e adolescentes com fibrose cística:
importância da imagem corporal e impacto do estado nutrional, idade e raça/cor na percepção dos pacientes e
responsáveis. Demetra: Alim, Nutr & Saúde 2018;13(3).
- Ribeiro JD, Fischer GB. Chronic obstructive pulmonary diseases in children. J Pediatr
2015;91:11-25.
- Silva Filho LVRF, Ferreira
FA, Reis FJC, MCA, Levy CE, Clark O et al. Infecção
por Pseudomonas aeruginosa
em pacientes com fibrose cística: evidências científicas sobre o impacto
clínico, diagnóstico e tratamento. J Bras Pneumol 2013;39(4):495-512.
- Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson R. Pseudomonas aeruginosa and other predictors of mortality and morbidity
in young children with cystic fibrosis. Pediatr Pulmonol
2002;34:91-100.
- Freitas FS, Parreira
VF, Ibiapina CC. Aplicação clínica do pico de fluxo da tosse: uma revisão de
literatura. Fisioter Mov 2010;23(3):495-502.
- Silverman EP, Carnaby-Mann G, Pitts T,
Davenport P, Okun MS, Sapienza
C. Concordance and discriminatory power of cough measurement devices for
individuals with parkinson
disease. Chest 2014;145(5):1089-96.
- França DC, Camargos PAM, Vieira BSPP, Pereira DAG, Parreira VF. Pico
de fluxo da tosse em pré-escolares: taxa de sucesso e reprodutibilidade teste-reteste. Fisioter Pesqui
2015;22(3):275-9.
- Batrawy SE, Elassal G. Is there a role for cough peak flow in
assessment of patients with severe COPD? Egypt J Chest Dis Tuberc
2014;1-5.
- Tzani P, Chiesa
S, Aiello M, Scarascia A, Catellani
C, Elia D. The value of cough peak flow in the
assessment of cough efficacy in neuromuscular patients: a cross sectional study. Eur J Phys Rehab Med 2014;50(4):427-32.
- Vilozni D, Lavi M, Ofek M, Sarouk I, Efrati O. Cough
characteristics and FVC maneuver in cystic fibrosis. Respir
Care 2014;59(12):1912-7.
- Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N et al. Diagnosis
of cystic fibrosis: consensus guidelines from the Cystic Fibrosis
Foundation. J
Pediatr 2017;181:S4-S15.
- Athanazio RA, Filho LVRFS,
Vergara AA, Ribeiro AF, Riedi CA, Procianoy
EFA et al. Diretrizes brasileiras de diagnóstico e
tratamento da fibrose cística. J Bras Pneumol 2017;43(3):219-45.
- Rodrigues JC, Cardieri JMA, Bussamra MHCF, Nakaie CMA, Almeida MB, Silva Filho LVF et
al. Provas de função pulmonar em crianças e adolescentes. In: Sociedade
Brasileira de Pneumologia e Tisiologia. Diretrizes para testes de função
pulmonar. J Bras Pneumol 2002;28(3):1-238.
- Beydon N, Davis SD,
Lombardi E, Allen JL, Arets HGM, Aurora P, et al. An Official American
Thoracic Society/European Respiratory Society Statement: Pulmonary Function
Testing in Preschool Children. Am J Respir Crit Care Med 2007;175:1304-45.
- Bianchi C, Baiardi P. Cough peak flows:
standard values for children and adolescents. Am J Phys
Med Rehabil 2008;87:461-7.
- Turck D, Braegger
CP, Colombo C, Declercq D, Morton
A, Pancheva R et al. ESPEN-ESPGHAN-ECFS
guidelines on nutrition care for infants, children, and adults with cystic
fibrosis. Clin Nutr 2016;35(3):557-77.
- Callegari J, Sidia M. Bioestatística: Princípios e Aplicações. Porto Alegre: Artmed;
2003;90.
- Moore JE, Mastoridis P. Clinical implications
of Pseudomonas aeruginosa location in the lungs of
patients with cystic fibrosis. J Clin Pharm Ther 2017;1-9.
- Reece E, Segurado R ,
Jackson A, McClean S, Renwick J, Greally P. Co-colonisation
with Aspergillus fumigatus and Pseudomonas
aeruginosa is associated with poorer health in
cystic fibrosis patients: an Irish registry analysis. BMC Pulm
Med 2017;17(1):70.
- Cantin AM, Hartl D, Konstan
MW, Chmiel JF.Inflammation
in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros 2015:14(4):419-30.
- Adler FR, Liou TG. The
dynamics of disease progression in cystic fibrosis. PloS One 2016;11(6):e0156752.
- Roberts AM, Pereira CL, Carby MR, Simon AR, Drey NS, Reed AK. The relationship between peak cough flow
and respiratory function testing (spirometry), and
the factors that influence this, post bilateral sequential single lung
transplantation: a cross-sectional feasibility study at a single centre cardiothoracic transplantation unit. J Heart
Lung Transplant 2018;37(4):S296.
- Feinstein AJ, Zhang Z, Chhetri DK, Long J.
Measurement of cough aerodynamics in healthy adults. Annal Otol Rhinol Laryngol 2017;126(5):396-400.
- Stoltz DA, Meyerholz
DK, Welsh MJ. Origins of cystic fibrosis lung disease. N
Engl J Med 2015;372(4):351-62.
- Kerem E. Cystic fibrosis: Priorities
and progress for future therapies. Paediatr Respir Rev 2017;24:14-16.
http://doi: 10.1016/j.prrv.2017.06.004.
- Button BM. Wilson C, Dentice R, Cox NS,
Middleton A, Tannenbaum, et al. Physiotherapy for
cystic fibrosis in Australia and New Zealand: A clinical practice guideline. Respirology 2016;21(4):656-67.
- Balfour-Lynn I. Royal Brompton Hospital Paediatric Cystic Fibrosis Team. Royal Brompton Hospital.
Clinical guidelines: Care of children with cystic fibrosis. 7.Ed. 2017. [citado
2018 Mar 16]. Disponível em:
<http://www.rbht.nhs.uk/healthprofessionals/clinical-departments/cystic-fibrosis/clinical-cf-guidelines-care-of-children/>
- Fracon JF, Sologuren MJJ, Gastaldi AC, Baraúna MA. Avaliação de crianças
com fibrose cística após 12 meses de fisioterapia respiratória. Fisioter Bras 2001;2(5):289-94.
- Torri BG, Barros RJ,
Oliveira AQ, Souza NS, Fernandes ABS. O método Pilates
melhora a função pulmonar e a mobilidade torácica de pacientes com doença
pulmonar obstrutiva crônica. Fisioter Bras 2017;18(1):56-62
- Harness-Brumley CL, Elliott AC, Rosenbluth DB, Raghavan D, Jain
R. Gender differences in outcomes of patients with cystic fibrosis. J Women
Health 2014;23(12):1012-20.
- Bonfim IM, Córdova ELM, Garcia CCB, Paro FM. Perfil dos pacientes com
fibrose cística atendidos no centro de referência
pediátrico do Espírito Santo. Rev Bras
Pesq Saúde 2018 (in press).
- Hauschild DB, Rosa AF, Ventura
JC, Barbosa E, Moreira EAM, Ludwig Neto N et al. Association of nutritional status with lung function and morbidity in
children and adolescents with cystic fibrosis: a 36-month cohort
study. Rev Paul Pediatr 2018; (Ahead of print)
- Konstan MV, Butler SM, Wohl MEB, Stoddard M, Matousek R,
Wagener JS et al. Growth and nutricional indexes in
early life predict pulmonary function in cystic fibrosis. J Pediatr
2003;142:624-30.
- Kulnik ST, Birring SS, Hodsoll J, Moxham J, Rafferty GF,
Kalra L. Higher cough flow is associated with lower
risk of pneumonia in acute stroke. Thorax 2016;71(5):474-5.
- Kulnik ST, MacBean
V, Birring SS, Moxham J, Rafferty GF, Kalra L. Accuracy of portable devices in measuring peak
cough flow. Physiol Meas 2015;36:243-57.
- Grupo Brasileiro de
Estudos de Fibrose Cística (GBEFC). Registro Brasileiro de Fibrose Cística
(REBRAFC) 2015. [citado 2018 Jul 3].
Disponível em: http://portalgbefc.org.br/relatorios-anuais-rebrafc/.